Western Blot: Technique, Theory, and Trouble Shooting
Abstract
Western blotting is a crucial method utilized in cell and molecular biology. By utilizing a western blot, researchers are capable of establish particular proteins from a fancy combination of proteins extracted from cells. The method makes use of three parts to perform this process: (1) separation by measurement, (2) switch to a strong assist, and (3) marking goal protein utilizing a correct major and secondary antibody to visualise. This paper will try to clarify the method and idea behind western blot, and provide some methods to troubleshoot.Keywords: Bio-medical analysis, protein, western blotGo to:
Introduction
Western blot is usually utilized in analysis to separate and establish proteins. In this system a mix of proteins is separated primarily based on molecular weight, and thus by kind, by means of gel electrophoresis. These outcomes are then transferred to a membrane producing a band for every protein. The membrane is then incubated with labels antibodies particular to the protein of curiosity.
The unbound antibody is washed off leaving solely the certain antibody to the protein of curiosity. The certain antibodies are then detected by growing the movie. As the antibodies solely bind to the protein of curiosity, just one band needs to be seen. The thickness of the band corresponds to the quantity of protein current; thus doing an ordinary can point out the quantity of protein current. The paper will first describe the protocol for western blot, accompanied by footage to assist the reader and idea to rationalize the protocol. This can be adopted by the theoretical clarification of the process, and within the later part, troubleshooting suggestions for widespread issues.
Technique
Cell lysis to extract protein
Protein might be extracted from totally different form of samples, reminiscent of tissue or cells. Below is the protocol to extract proteins from adherent cells.
Adherent cells:
- Wash cells within the tissue tradition flask or dish by including chilly phosphate buffered saline (PBS) and rocking gently. Discard PBS. (Tip: Keep tissue tradition dish on ice all through).
- Add PBS and use a cell scraper to dislodge the cells. Pipette the combination into microcentrifuge tubes.
- Centrifuge at 1500 RPM for five minutes and discard the supernatant.
- Add 180 μL of ice chilly cell lysis buffer with 20 μL contemporary protease inhibitor cocktail. (Tip: If protein focus just isn’t excessive sufficient on the finish, it’s suggested to repeat the process with a better proportion of protease inhibitor cocktail).
- Incubate for 30 minutes on ice, and then make clear the lysate by spinning for 10 minutes at 12,000 RPM, at 4°C.
- Transfer supernatant (or protein combine) to a contemporary tube and retailer on ice or frozen at -20°C or -80°C.
- Measure the focus of protein utilizing a spectrophotometer.
Sample preparation
 decide the amount of protein extract to make sure 50 μg in every effectively.
decide the amount of protein extract to make sure 50 μg in every effectively.- Add 5 μL pattern buffer to the pattern, and make the amount in every lane equalized utilizing double distilled H2O (dd H2O). Mix effectively. (Tip: Total quantity of 15 μL per lane is usually recommended).
- Heat the samples with dry plate for five minutes at 100°C.
Gel preparation
- After making ready the 10% stacking gel answer, assemble the rack for gel solidification [Figure 1]. (Tip: 10% AP and TEMED solidify the answer; due to this fact, each gels might be ready on the identical time, if the abovementioned reagents usually are not added till the top).Figure 1Assembled rack for gel solidification
- Add stacking gel answer fastidiously till the extent is the same as the inexperienced bar holding the glass plates [Figure 2]. Add H2O to the highest. Wait for 15–30 minutes till the gel turning solidified. (Tip: Using a suction pipette could make the method of including the gel to the glass plate simpler).Open in a separate windowFigure 2Add gel answer utilizing a switch pipette
- Overlay the stacking gel with the separating gel, after eradicating the water. (Tip: It is healthier to tilt the equipment and use a paper towel to take away the water).
- Insert the comb, making certain that there aren’t any air bubbles.
- Wait till the gel is solidified. (Tip: Solidification might be simply checked by leaving some gel answer in a tube).
Electrophoresis
- Pour the working buffer into the electrophorator [Figure 3].Open in a separate windowFigure 3Add working buffer to the electrophorator
- Place gel contained in the electrophorator and connect with an influence provide. (Tip: When connecting to the ability supply all the time join crimson to crimson, and black to black).
- Make positive buffer covers the gel utterly, and take away the comb fastidiously.
- Load marker (6 μL) adopted by samples (15 μL) in to every effectively [Figure 4].Open in a separate windowFigure 4Add samples and molecular marker to the gel, after eradicating the combs
- Run the gel with low voltage (60 V) for separating gel; use greater voltage (140 V) for stacking gel [Figure [Figure5a5a and andbb].
 Figure 5(a) Samples working by means of the stacking gel (decrease voltage). (b): Samples working by means of the separating gel (greater voltage)
Figure 5(a) Samples working by means of the stacking gel (decrease voltage). (b): Samples working by means of the separating gel (greater voltage) - Run the gel for about an hour, or till the dye entrance runs off the underside of the gel [Figure 6].Figure 6Run the gel to the underside of the electrophorator
Electrotransfer
- Cut 6 filter sheets to suit the measurement of the gel, and one polyvinylidene fluoride (PDVF) membrane with the identical dimensions.
- Wet the sponge and filter paper in switch buffer, and moist the PDVF membrane in methanol.
- Separate glass plates and retrieve the gel.
- Create a switch sandwich as follows:Sponge3 Filter PapersGel PVDF3 Filter Papers(Tip: Ensure there aren’t any air bubbles between the gel and PVDF membrane, and squeeze out further liquid).
- Relocate the sandwich to the switch equipment, which needs to be positioned on ice to take care of 4°C. Add switch buffer to the equipment, and make sure that the sandwich is roofed with the buffer. Place electrodes on high of the sandwich, making certain that the PVDF membrane is between the gel and a optimistic electrode [Figure 7].Figure 7Transfer needs to be executed on ice
- Transfer for 90 minutes [Figure 8]. (Tip: The working time needs to be proportional to the thickness of the gel, so this can be diminished to 45 minutes for 0.75 mm gels).Open in a separate windowFigure 8Membrane after switch
Blocking and antibody incubation
- Block the membrane with 5% skim milk in TBST* for 1 hour.
- Add major antibody in 5% bovine serum albumin ( BSA) and incubate in a single day in 4°C on a shaker [Figure 9].Figure 9Use a shaker to incubate the membrane with antibody
- Wash the membrane with TBST for five minutes. Do this Three occasions. (Tip: All washing and antibody incubation steps needs to be executed on a shaker at room temperature to make sure even agitation).
- Add secondary antibody in 5% skim milk in TBST, and incubate for 1 hour.
- Wash the membrane with TBST for five minutes. Do this Three occasions
- Prepare ECL combine (following the proportion of answer A and B supplied by the producer). Incubate the membrane for 1–2 minutes [Figure 10]. (Tip: Use a 1000 μL pipette to make sure that ECL covers the highest and backside of the membrane).Figure 10Incubate the membrane with ECL combine utilizing a 1000 μL pipette to assist the method
- Visualize the end result in the dead of night room [Figure 11]. (Tip: If the background is simply too robust, cut back publicity time).Figure 11Use the cassette to reveal the membrane in the dead of night room
Recipe
- Dissolve the next in 800 ml of distilled H2O
- 8.Eight g of NaCl
- 0.2g of KCl
- 3g of Tris base
- Add 500ul of Tween-20
- Adjust the pH to 7.4
- Add distilled H2O to 1L
- Sterilize by filtration or autoclaving
Theory
Sample preparation
Cell lysates are the commonest type of pattern used for western blot. Protein extraction makes an attempt to gather all of the proteins within the cell cytosol. This needs to be executed in a chilly temperature with protease inhibitors to stop denaturing of the proteins. Since tissue pattern show a better diploma of construction, mechanical invention, reminiscent of homogenization, or sonication is required to extract the proteins.
After extracting the protein, it is extremely essential to have a good suggestion of the extract’s focus. This ultimately permits the researcher to make sure that the samples are being in contrast on an equal foundation. Protein focus is usually measured utilizing a spectrophotometer. Using this focus permits to measure the mass of the protein that’s being loaded into every effectively by the connection between focus, mass, and quantity.
After figuring out the suitable quantity of the pattern, it’s diluted right into a loading buffer, which comprises glycerol in order that the samples sink simply into the wells of the gel. A monitoring dye (bromophenol blue) can also be current within the buffer permitting the researcher to see how far the separation has progressed. The pattern is heated after being diluted right into a loading buffer, so as to denature the upper order construction, whereas retaining sulfide bridges. Denaturing the excessive construction ensures that the unfavorable cost of amino acids just isn’t neutralized, enabling the protein to maneuver in an electrical area (utilized throughout electrotransfer).
It can also be crucial to have optimistic and unfavorable controls for the pattern. For a optimistic management a identified supply of goal protein, reminiscent of purified protein or a management lysate is used. This helps to verify the identification of the protein, and the exercise of the antibody. A unfavorable management is a null cell line, reminiscent of β-actin, is used as effectively to verify that the staining just isn’t nonspecific.
Gel electrophoresis
Western blot makes use of two various kinds of agarose gel: stacking and separating gel. The greater, stacking gel is barely acidic (pH 6.8) and has a decrease acrylamide focus making a porous gel, which separates protein poorly however permits them to kind skinny, sharply outlined bands. The decrease gel, referred to as the separating, or resolving gel, is primary (pH 8.8), and has a better polyacrylamide content material, making the gel’s pores narrower. Protein is thus separated by their measurement extra so on this gel, because the smaller proteins to journey extra simply, and therefore quickly, than bigger proteins.
The proteins when loaded on the gel have a unfavorable cost, as they’ve been denatured by heating, and will journey towards the optimistic electrode when a voltage is utilized. Gels are often made by pouring them between two glass or plastic plates, utilizing the answer described within the protocol part. The samples and a marker are loaded into the wells, and the empty wells are loaded with pattern buffer. The gel is then related to the ability provide and allowed to run. The voltage is essential, as a excessive voltage can overheat and distort the bands.
Blotting
After separating the protein combination, it’s transferred to a membrane. The switch is completed utilizing an electrical area oriented perpendicular to the floor of the gel, inflicting proteins to maneuver out of the gel and onto the membrane. The membrane is positioned between the gel floor and the optimistic electrode in a sandwich. The sandwich features a fiber pad (sponge) at every finish, and filter papers to guard the gel and blotting membrane [Figure 12]. Here two issues are crucial: (1) the shut contact of gel and membrane to make sure a transparent picture and (2) the position of the membrane between the gel and the optimistic electrode. The membrane should be positioned as such, in order that the negatively charged proteins can migrate from the gel to the membrane. This kind of switch is named electrophoretic switch, and might be executed in semi-dry or moist situations. Wet situations are often extra dependable as it’s much less more likely to dry out the gel, and is most popular for bigger proteins.Figure 12
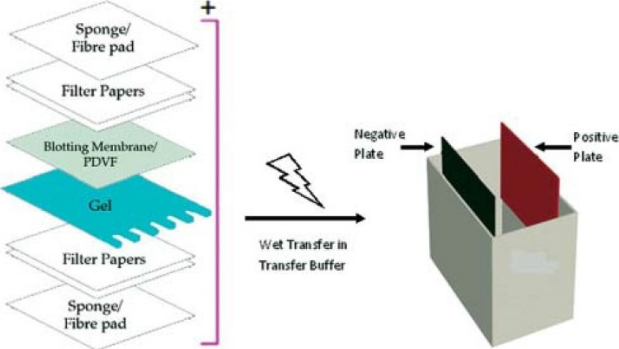
Assembly of a sandwich in western Blot
The membrane, the strong assist, is a vital a part of this course of. There are two sorts of membrane: nitrocellulose and PVDF. Nitrocellulose is used for its excessive affinity for protein and its retention skills. However, it’s brittle, and doesn’t enable the membrane for use for reprobing. In this regard, PVDF membranes present higher mechanical assist and enable the blot to be reprobed and saved. However, the background is greater within the PVDF membranes and due to this fact, washing fastidiously is essential.
Washing, blocking and antibody incubation
Blocking is a vital step of western blotting, because it prevents antibodies from binding to the membrane nonspecifically. Blocking is usually made with 5% BSA or nonfat dried milk diluted in TBST to scale back the background.
Nonfat dried milk is usually most popular as it’s cheap and extensively obtainable. However, milk proteins usually are not suitable with all detection labels, so care should be taken to decide on the suitable blocking answer. For instance, BSA blocking options are most popular with biotin and AP antibody labels, and antiphosphoprotein antibodies, since milk comprises casein, which is itself a phosphoprotein and biotin, thus interfering with the assay outcomes. It is usually a very good technique to incubate the first antibody with BSA since it’s often wanted in greater quantities than the secondary antibody. Putting it in BSA answer permits the antibody to be reused, if the blot doesn’t give good end result.
The focus of the antibody relies on the instruction by the producer. The antibody might be diluted in a wash buffer, reminiscent of PBS or TBST. Washing is essential because it minimized background and removes unbound antibody. However, the membrane shouldn’t be left to clean for a very very long time, as it may well additionally cut back the sign.
The membrane is then detected utilizing the label antibody, often with an enzyme reminiscent of horseradish peroxidase (HRP), which is detected by the sign it produces similar to the place of the goal protein. This sign is captured on a movie which is often developed in a darkish room.
Quantification
It is essential to remember that the info produced with a western blot is often thought of to be semi-quantitative. This is as a result of it supplies a relative comparability of protein ranges, however not an absolute measure of amount. There are two causes for this; first, there are variations in loading and switch charges between the samples in separate lanes that are totally different on separate blots. These variations will must be standardized earlier than a extra exact comparability might be made. Second, the sign generated by detection just isn’t linear throughout the focus vary of samples. Thus, because the sign produced just isn’t linear, it shouldn’t be used to mannequin the focus.
Troubleshooting
Even although the process for western blot is straightforward, many issues can come up, resulting in sudden outcomes. The drawback might be grouped into 5 classes: (1) uncommon or sudden bands, (2) no bands, (3) faint bands or weak sign, (4) excessive background on the blot, and (5) patchy or uneven spots on the blot.
Unusual or sudden bands might be resulting from protease degradation, which produces bands at sudden positions. In this case it’s advisable to make use of a contemporary pattern which had been stored on ice or alter the antibody. If the protein appears to be in too excessive of a place, then reheating the pattern might help to interrupt the quaternary protein construction. Similarly, blurry bands are sometimes attributable to excessive voltage or air bubbles current throughout switch. In this case, it needs to be ensured that the gel is run at a decrease voltage, and that the switch sandwich is ready correctly. In addition, altering the working buffer may also assist the issue. Nonflat bands might be the results of too quick of a journey by means of the gel, resulting from low resistance. To repair this the gel needs to be optimized to suit the pattern. Finally, white (unfavorable) bands on the movie are resulting from an excessive amount of protein or antibody.
Another drawback: no bands may also come up resulting from many causes associated to antibody, antigen, or buffer used. If an improper antibody is used, both major or secondary, the band is not going to present. In addition, the focus of the antibody needs to be acceptable as effectively; if the focus is simply too low, the sign will not be seen. It is essential to keep in mind that some antibodies usually are not for use for western blot. Another purpose for no seen bands is the bottom focus or absence of the antigen. In this case, antigen from one other supply can be utilized to verify whether or not the issue lies with the pattern or with different parts, such because the antibody. Moreover, extended washing may also lower the sign. Buffers may also contribute to the issue. It needs to be ensured that buffers just like the switch buffer, TBST, working buffer and ECL are all new and noncontaminated. If the buffers are contaminated with sodium azide, it may well inactivate HRP.
Similarly, weak indicators might be attributable to low focus of antibody or antigen. Increasing publicity time may also assist to make the band clearer. Another purpose might be nonfat dry milk masking the antigen. In this case use BSA or lower the quantity of milk used.
High background is usually attributable to too excessive focus of the antibody, which might bind to PVDF membranes. Another drawback might be the buffers, which can be too previous. Increasing the washing time may also assist to lower the background. Additionally, too excessive of an publicity may also result in this drawback. Therefore, it’s advisable to verify totally different publicity occasions to attain an optimum time.
Patchy and uneven spots on the blot are often attributable to improper switch. If there are air bubbles trapped between the gel and the membrane, it’ll seem darker on the movie. It can also be essential to make use of a shaker for all incubation, in order that there is no such thing as a uneven agitation through the incubation. Once once more, washing is of utmost significance as effectively to clean the background. This drawback may also be attributable to antibodies binding to the blocking brokers; on this case one other blocking agent needs to be tried. Filtering the blocking agent may also assist to take away some contaminants. Finally, this drawback may also be attributable to aggregation of the secondary antibody; on this case, the secondary antibody needs to be centrifuged and filtered to take away the aggregated.Go to:
Conclusion
Western blot is a way that could be very helpful for protein detection because it permits the consumer to quantify the protein expression as effectively. This paper coated the protocol, the idea behind that protocol, and some troubleshooting methods. Western blot might be seen as an intricate stability, because the researcher makes an attempt to get a nonspecific, but robust sign.